Scripps Research scientists have identified new proteins that may be targeted by upcoming cancer drugs, using a dual protein-mapping approach.
Effectively combating cancer often entails halting the multiplication of cancer cells, which depends on understanding the proteins these cells need to survive. Protein profiling is crucial in this effort, as it enables researchers to pinpoint the proteins—and their specific components—that should be targeted by future drugs. However, previous methods, when used alone, have lacked the precision necessary to identify all possible protein targets, resulting in some being overlooked.
Now, by combining two methods of protein analysis, a team of chemists at Scripps Research has mapped more than 300 small molecule-reactive cancer proteins, as well as their small molecule binding sites. Revealing key protein targets that, when disrupted with certain chemical compounds (or small molecules), halt cancer cell growth may eventually enable the development of more effective and precise cancer treatments. The findings were published in Nature Chemistry on August 13, 2024.
Techniques in Protein Profiling
“One method gave us a broad view of which proteins were interacting with the chemicals, and the second method showed exactly where those interactions were happening,” says co-senior author Benjamin Cravatt, PhD, the Norton B. Gilula Chair in Biology and Chemistry at Scripps Research.
Both methods are forms of activity-based protein profiling (ABPP), a technique that Cravatt pioneered to capture protein activity on a global scale. The research team used their dual approach to flag both the proteins and protein sites that interacted with a library of stereoprobes—chemical compounds designed to permanently bind to proteins in a selective manner. Stereoprobes are used to study protein functions and identify possible drug targets.
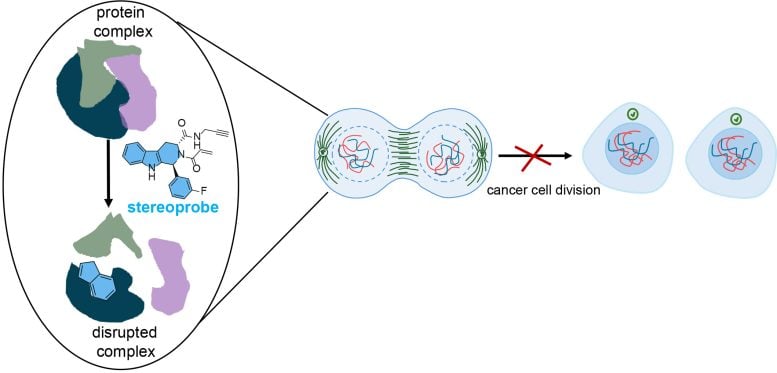
“We made a conscious effort to design our stereoprobes with chemical features that tend to be underrepresented in compounds typically used in drug discovery settings,” says co-senior author Bruno Melillo, PhD, an institute investigator in the Department of Chemistry at Scripps Research. “This strategy increases our chances of making discoveries that can advance biology, and eventually translate into improvements to human health.”
The research team’s stereoprobes were electrophilic, meaning they were designed to irreversibly bind to proteins—specifically to cysteine. This amino acid is pervasive in proteins, including those found in cancer cells, and it helps form important structural bonds. When chemicals react with cysteine, they can disrupt these bonds and cause proteins to malfunction, which interferes with cell growth, and many cancer drugs irreversibly bind to cysteines on proteins.
“We also focused on cysteine because it’s the most nucleophilic amino acid,” says first author Evert Njomen, PhD, an HHMI Hanna H. Gray Fellow at Scripps Research and a postdoctoral research associate in Cravatt’s lab.
Detailed Analysis Using ABPP
To find out which specific proteins would bond with the stereoprobes, the team turned to a method known as protein-directed ABPP. Using this approach, the researchers uncovered more than 300 individual proteins that reacted with the stereoprobe compounds. But still, they wanted to dig deeper and identify the reactions’ precise locations.
The second method, called cysteine-directed ABPP, pinpointed exactly where the stereoprobes were binding on the proteins. This allowed the team to “zoom in” on a specific protein pocket and examine whether the cysteine within reacted with the stereoprobes, similar to focusing on a single spot on a puzzle board to see if a particular piece fits.
Each stereoprobe molecule had two main components: the binding part and the electrophilic part. Once the binding component recognizes the cancer cell protein pocket, hopefully, the stereoprobe molecule can enter—like how a key needs to fit in a lock. When a stereoprobe remained in a pocket that’s critical to the cancer cell’s function, it blocked the protein from binding to other proteins—ultimately preventing cell division.
“By targeting these very specific stages in the cell cycle, there’s potential to slow down the growth of cancer cells,” says Njomen. “A cancer cell would stay in what is almost a state of two cells, and your body’s immune system would detect it as defective and direct it to die.”
Identifying precise protein regions that are critical to cancer cell survival could help researchers develop more targeted treatments to stop cells from multiplying.
Among the team’s other key findings was confirming that their double-pronged approach painted a more accurate picture of protein-stereoprobe reactivity than a single method.
“We’ve always known that both methods had their drawbacks, but we didn’t know exactly how much information was lost by using just one technique,” says Njomen. “It was surprising to see that a substantial number of protein targets were missed when we used one platform over the other.”
The team hopes that their findings will one day inform new cancer therapies targeting cell division. In the meantime, Njomen wants to design new stereoprobe libraries to uncover protein pockets implicated in illnesses beyond cancer, including inflammatory disorders.
“Many proteins have been implicated in diseases, but we don’t have stereoprobes to research them,” she said. “Moving forward, I’d like to find more protein pockets that we can study for drug discovery purposes.”
Reference: “Multi-tiered chemical proteomic maps of tryptoline acrylamide–protein interactions in cancer cells” by Evert Njomen, Rachel E. Hayward, Kristen E. DeMeester, Daisuke Ogasawara, Melissa M. Dix, Tracey Nguyen, Paige Ashby, Gabriel M. Simon, Stuart L. Schreiber, Bruno Melillo and Benjamin F. Cravatt, 13 August 2024, Nature Chemistry.
DOI: 10.1038/s41557-024-01601-1
This work and the researchers involved were supported by funding from the National Institutes of Health (U19 AI142784 and R35 CA231991); Cancer Research UK (CGCATF-2021/100012 and CGCATF-2021/100021) the National Cancer Institute (OT2CA278688 and OT2CA278692); the Howard Hughes Medical Institute Hanna H. Gray Fellowship (NGT15176), the Jane Coffin Childs Memorial Fellowship, and Vividion Therapeutics.