
A research team from the LKS Faculty of Medicine, the University of Hong Kong (HKUMed) has developed a new way to break through the current limited throughput in optimizing precise genome editors at scale, and engineer hundreds (or more) of base editor variants in parallel instead of via current laborious one-at-a-time testing, and inform users of the most suitable ones for therapeutic genome editing. The finding has been published in Cell Systems and a patent application has been filed based on this work.
Base editing is a newer CRISPR-based genome editing technology, and emerges to be a safer tool for tackling genetic diseases with single-base mutations (such as sickle cell disease, familial hypercholesterolemia, etc.) in DNA by correcting them to their normal form.
However, existing base editing can result in different outcomes depending on the type and version of the base editor used, the sequence composition of the target DNA, and the position of the DNA base(s) to-be-converted. Picking a sub-optimal base editor for application can generate incorrect edits and extra mutations around the target DNA base, which could cause undesired effects.
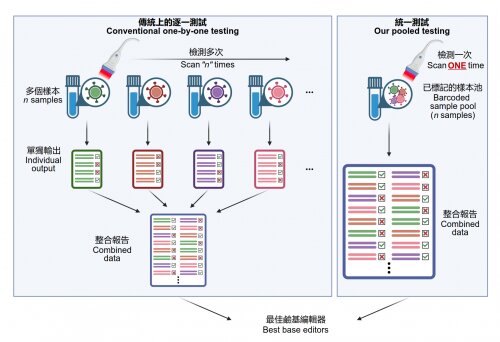
Currently, laborious exhaustive one-at-a-time testing has to be done to characterize the editing performance of the tens of available base editors to optimize their use on each therapeutic locus. In addition, many therapeutic loci do not yet have an existing optimized base editor for precise editing. Despite efforts worldwide, creating a new base editor can take months or years using conventional methods.
HKUMed’s research team has successfully developed a platform coupling a base editor reporter system with CombiSEAL, a state-of-the-art technology previously developed to quickly engineer hundreds (or more) of base editor variants in parallel with combinations of varying enzymatic deaminase domains and CRISPR/ Cas9-based DNA-recognition domains. The compatibility and performance of these variants had not yet been characterized and compared in a head-to-head manner.
The team applied the platform to quantitatively readout each variant’s editing efficiency, purity, sequence motif preference, and bias in generating single and multiple base conversions in human cells, which helps select the most suitable ones for therapeutic target by generating a particular type of base conversion with maximal efficiency and minimal undesired edits.
The team extended the use of the platform to further enhance the efficiency of the current base editor system. The team members performed a screen focusing on engineering the stem-loop-2 region of the sgRNA (a single guide RNA) scaffold used in the base editor system, and successfully identified two novel sgRNA scaffold variants, SV48 and SV240, that outperformed the wild-type scaffold to achieve greater (up to 2.2-fold higher) base editing efficiency.
Furthermore, the team demonstrated that the platform could not only be used for base editor characterization and screening, but also is compatible with other precise genome editor systems such as prime editors. This could expand the scope of the search for other suitable editors to correct genetic mutations at therapeutic targets where a base editor is not applicable.
This platform is powerful in accelerating the engineering of next-generation precise genome editors and the adaptation of these editors in future therapeutic uses.
“It is like an accelerated checkout process in stores. Since all product items (i.e. base editor variants) are tagged with a barcode, when it comes to the checkout counter barcode scanner, we need to only put all items in bulk into the basket at the checkout counter. The scanner can automatically identify all items and complete the payment (i.e. base editing performance analysis in our case). There is no need to individually test each base editor one-by-one,” Dr. Alan Wong Siu-lun, Associate Professor of the School of Biomedical Sciences, HKUMed, explained.
The research was led by Dr. Alan Wong Siu-lun, Associate Professor, School of Biomedical Sciences, HKUMed. John Fong Hoi-chun, Ph.D. student, was first author, with assistance from Dr. Chu Hoi-yee and Dr. Zhou Peng, postdoctoral fellows, School of Biomedical Sciences, HKUMed.
More information:
John H.C. Fong et al, Parallel engineering and activity profiling of a base editor system, Cell Systems (2023). DOI: 10.1016/j.cels.2023.03.007
Provided by
The University of Hong Kong
Citation:
New platform slashes time to engineer and select the best genome editors for specific applications (2023, June 29)
retrieved 29 June 2023
from https://phys.org/news/2023-06-platform-slashes-genome-editors-specific.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.