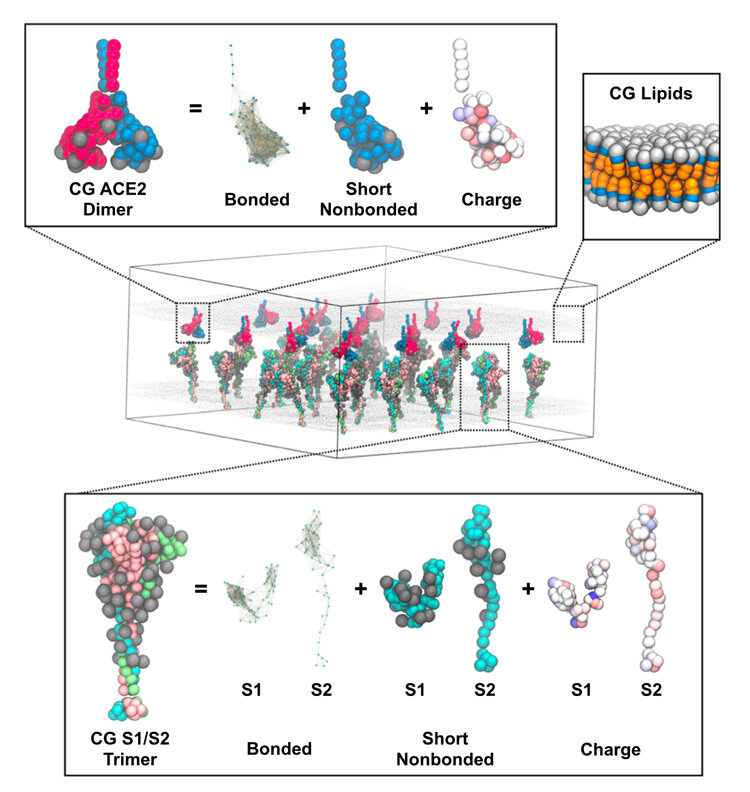
 simulation of spike trimers in membrane interacting with an adjacent membrane with ACE2 dimers. The insets depict the CG model components for the spike trimer (bottom), ACE2 dimer (upper left), and lipid membrane (upper right). Credit: Pak, A.J., Yu, A., Ke, Z. et al.")
The mystery of exactly how the SARS-CoV-2 virus infects human lung cells remains largely hidden to experimental scientists. Now, however, the devilish details of the mechanism by which the coronavirus fuses to host cells has been suggested through simulations by University of Chicago researchers using the Frontera supercomputer at the Texas Advanced Computing Center (TACC).
The computer models show the cooperative behavior of host cell receptor proteins that leads to their own infection. The work can be applied to help understanding the increased virulence of coronavirus variants such as delta, omicron, and more.
“We discovered that the spike protein interacts with two ACE2 receptors in a very cooperative fashion,” said Gregory Voth, a distinguished professor of chemistry at the University of Chicago. “This is a fundamental biophysical insight.”
Voth is senior author on the study that modeled the coronavirus and receptor cell interactions with computer simulations published in the journal Nature Communications in February 2022.
Like a soccer ball with spikes, the spike proteins adorn the surface of the coronavirus. The spikes seek out and fuse with the angiotensin-converting enzyme 2 (ACE2) protein receptors in human lung cells. The spike protein is composed of two main parts. The S1 domain contains the receptor binding domain that recognizes ACE2 proteins. And the S2 domain contains the fusion machinery, which is protected and covered like a husk by the S1 domain.
The simulations reveal how one ACE2 receptor protein holds on to the coronavirus spike and weakens it while the other starts pulling it apart. The S1 domain then falls apart and exposes the fusion machinery. This “one-two” punch primes the virus for fusion and entry into human lung host cells.
“It appears that variants such as delta and omicron may accentuate that behavior even more—it’s a key step. Ultimately, future antibodies and possibly molecular pharmaceuticals should be able to interfere with this process,” Voth said.
Voth and colleagues developed what they call “bottom-up coarse-grained models” that took cryo-electron tomography data from the lab of study co-author John Briggs of the Max Planck Institute of Biochemistry. They combined it with atomistic molecular dynamics simulations. The data generated fed into a theoretical framework that developed the coarse-grained models.
“The coarse-grained models are up to 1,000 times faster than straight-up atomistic molecular dynamics simulations, but they retain the essential physical features,” Voth said. This method provides an enormous savings in time and money on the computations.
The science team was awarded supercomputer resources and services by the COVID-19 HPC Consortium, a public-private effort in support of COVID-19 research. Through the consortium, they used the National Science Foundation-funded Frontera system at TACC; the Witherspoon computer cluster at IBM Research; and resources of the Oak Ridge Leadership Computing Facility at the Oak Ridge National Laboratory.
“We computed all-atom molecular dynamics data on Frontera and used analysis tools available from TACC—both were very valuable,” Voth said.
Voth’s team submitted their paper before the delta and omicron variants were known, and therefore didn’t predict the mutations. But they did go back and revise the models to investigate the variants.
“Delta has something like an opening in the spike that happens more readily than in prior coronavirus mutations,” Voth said. “It felt exciting from a scientific view to see behaviors that hadn’t been seen before.”
Voth referred to cryo-electron microscopy lab data showing the structure of a soluble spike protein with two ACE2 receptors bound to it. But he distinguished this crystallized example from what he investigated using simulations in the more realistic environment of many proteins interacting with each other on membrane sheets.
“Supercomputers, if used well and based on good physics, can provide a whole new way of looking at these processes. Through computer simulation, one can study things that can’t presently be done with experiments. Simulation and experiments work very well together, hand in hand,” Voth said.
First complete coronavirus model shows cooperation
Alexander J. Pak et al, Cooperative multivalent receptor binding promotes exposure of the SARS-CoV-2 fusion machinery core, Nature Communications (2022). DOI: 10.1038/s41467-022-28654-5
Texas Advanced Computing Center
Citation:
Supercomputer simulations reveal the details of coronavirus fusion (2022, May 6)
retrieved 6 May 2022
from https://phys.org/news/2022-05-supercomputer-simulations-reveal-coronavirus-fusion.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.