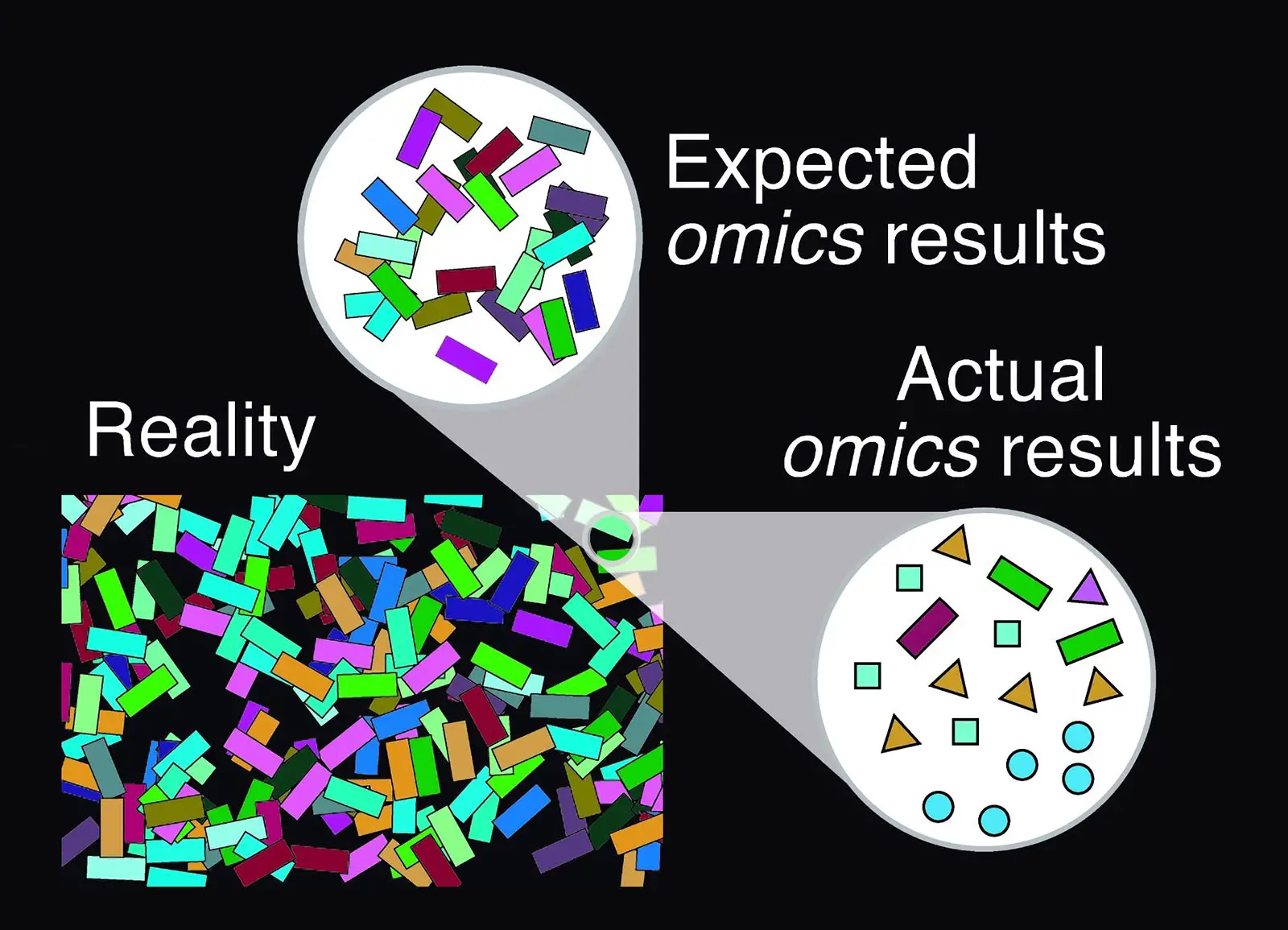
Shortcomings of current metagenomic analyses. Credit: Serrano-Antón et al., CC-BY 4.0
Research study of simulated microbial communities shows analyses are flawed by incomplete DNA databases.
Common approaches to analyzing DNA from a community of microbes, called a microbiome, can yield erroneous results, in large part due to the incomplete databases used to identify microbial DNA sequences. A team led by Aiese Cigliano of Sequentia Biotech SL, and Clemente Fernandez Arias and Federica Bertocchini of the Centro de Investigaciones Biologicas Margarita Salas, report these findings in a research paper published on February 8 in the open-access journal PLOS ONE.
Microbiomes have been the focus of intense research efforts in recent decades. These studies range from attempts to understand conditions such as obesity and autism by examining the human gut, to finding microbes that degrade toxic compounds or produce biofuels by studying environmental communities. The most commonly used methods for studying microbial communities rely on comparing the DNA obtained from a biological sample to sequences in genome databanks. Therefore, researchers can only identify DNA sequences that are already in the databases – a fact that may severely compromise the reliability of microbiome data in unexpected ways.
To test the consistency of current methods of microbiome analysis, researchers used computer simulations to create virtual microbiome communities that imitate real-world bacterial populations. They used standard techniques to analyze the virtual communities and compared the results with the original composition. The experiment showed that results from DNA analyses can bear little resemblance to the actual composition of the community, and that a large number of the species “detected” by the analysis are not actually present in the community.
For the first time, the study demonstrates significant flaws in the techniques currently used to identify microbial communities. The researchers conclude that there is a need for increased efforts to collect genome information from microbes and to make that information available in public databases to improve the accuracy of microbiome analysis. In the meantime, the results of microbiome studies should be interpreted with caution, especially in cases where the available genomic information from those environments is still scarce.
The authors add: “This study reveals intrinsic constraints in metagenomic analysis stemming from current database limitations and how genomic information is used. To enhance the reliability of metagenomic data, a research effort is necessary to improve both database contents and analysis methods. Meanwhile, metagenomic data should be approached with great care.”
Reference: “The virtual microbiome: A computational framework to evaluate microbiome analyses” by Belén Serrano-Antón, Francisco Rodríguez-Ventura, Pere Colomer-Vidal, Riccardo Aiese Cigliano, Clemente F. Arias and Federica Bertocchini, 8 February 2023, PLOS ONE.
DOI: 10.1371/journal.pone.0280391
Funding: FB and CFA gratefully acknowledge support by the Roechling foundation. BS was partially supported by MINECO grant MTM2017-85020-P. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.