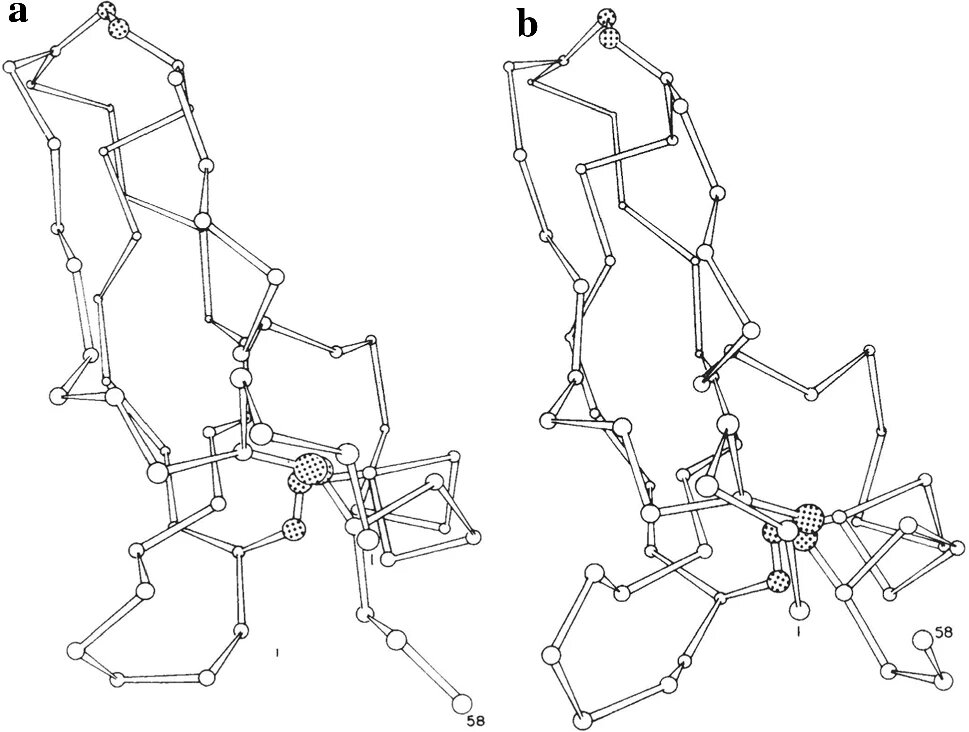
 X-ray original structure; b) time-evolved structure after 3.2 ps. In both drawings, alpha carbons are indicated by circles, while sulfides in disulfide bonds are represented by dashed circles. Image taken from McCammon et al. (1977), on p. 586. Credit: <i>The European Physical Journal H</i> (2022). DOI: 10.1140/epjh/s13129-022-00043-y")
Life is motion. And so, to understand how living organisms function, one must understand the movement and reorganization of the atoms and molecules that compose them. The approach called “molecular dynamics simulation” enables scientists to use computer programs to simulate the dynamic motion of all the atoms in a molecular system as a function of time.
In a new paper inEPJ Historical Perspectives on Contemporary Physics, Daniele Macuglia from Peking University in Beijing, China, Benoît Roux from the University of Chicago in Chicago, U.S., and Giovanni Ciccotti from the University of Rome in Rome, Italy, explain how the theoretical chemist Martin Karplus and his team carried out the first molecular dynamics simulation of a large biological molecule, a protein, deeply impacting biology and the physical sciences in the 20thand 21st centuries.
Currently, machine learning researchers are using biomolecular simulations to better understand their time-dependent motions and the function that governs the forces between them.
In the early 1970s, physicists and physical chemists started to use molecular dynamics simulation to study the behavior of simple substances such as water and liquids formed from noble gases. Martin Karplus and his team took this method further by applying it for the first time to a large biological molecule—a protein.
Proteins can be thought of as miniature machines whose function arises, in part, from the way they fold and contort into different shapes over time. Because of their complexity, simulating their changes over time was a particular challenge.
Karplus first publicized this approach with a 1977 paper showing the first molecular dynamics simulation of a protein. More recently, the deep impact of his contribution to accurately modeling chemical reactions was acknowledged through being awarded the 2013 Nobel Prize in Chemistry together with Arieh Warshel and Michael Levitt.
The authors conclude that Karplus’s 1977 article opened the door to pursue a line of research that led him to successfully merge computational statistical mechanics with biochemistry.
More information:
Daniele Macuglia et al, The emergence of protein dynamics simulations: how computational statistical mechanics met biochemistry, EPJ Historical Perspectives on Contemporary Physics (2022). DOI: 10.1140/epjh/s13129-022-00043-y
Citation:
Legacy of a molecular dynamics trailblazer: Computer simulations meet biochemistry (2022, November 17)
retrieved 17 November 2022
from https://phys.org/news/2022-11-legacy-molecular-dynamics-trailblazer-simulations.html
This document is subject to copyright. Apart from any fair dealing for the purpose of private study or research, no
part may be reproduced without the written permission. The content is provided for information purposes only.